


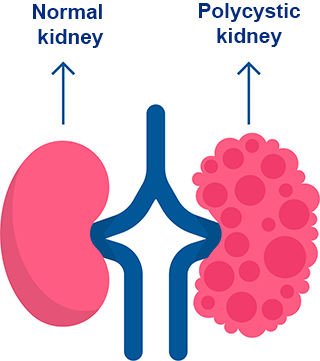
Polycystic kidney disease (PKD) is a genetic disorder characterized by the formation of fluid-filled cysts in the kidneys.
The presence and growth of these cysts can cause the kidneys to enlarge, leading to compromised kidney function over time.
PKD is classified as a ciliopathy, which is a group of genetic disorders caused by dysfunction of cilia—small, hair-like structures on the surface of many types of cells, including kidney cells. In PKD, abnormal ciliary function contributes to the formation of kidney cysts.
This condition is the most common genetic cause of kidney failure worldwide.
Polycystic kidney disease (PKD) is a genetic disorder caused by mutations in one of two genes:
In approximately 90% of cases, PKD is inherited, meaning it is passed down from a parent. However, in about 10% of cases, the disease arises spontaneously due to a de novo mutation (a new mutation that occurs in the individual without being inherited).
Since PKD is a genetic condition, it is neither infectious nor contagious.
There are two main types of polycystic kidney disease:
Autosomal dominant polycystic kidney disease (ADPKD): This form affects approximately 1 to 2 people per 1,000 in the general population.
Autosomal recessive polycystic kidney disease (ARPKD): This rare form affects approximately 1 to 2 people per 40,000 individuals.
The onset of symptoms varies, with most cases appearing in early to mid-adulthood. However, in some instances, the disease is only discovered postmortem during an autopsy, or it may manifest in childhood.
In approximately 50% of cases, cysts are also present in the liver.
Symptoms are often related to the effects of kidney cysts and may include:
Early in the disease, kidney dysfunction can also cause high blood pressure (hypertension), anemia and urinary infections.
Additionally, brain aneurysms are associated with approximately 10-20% of cases, and around 15% of individuals with PKD experience subarachnoid hemorrhages. High blood pressure (hypertension) is present in about 50% of patients at the time of diagnosis.
The diagnosis of polycystic kidney disease (PKD) is based on clinical signs and symptoms, family history, imaging studies and genetic and laboratory tests (blood and urine analyses).
In advanced cases, where the kidneys are significantly enlarged and palpable, the diagnosis may be apparent.
The following imaging techniques are commonly used:
Urine analysis may show mild proteinuria (protein in the urine). Hematuria (blood in the urine) can occur in varying degrees, but red blood cell casts are rare. Pyuria (pus in the urine) is common, even in the absence of bacterial infection. Occasionally, gross hematuria is observed, often due to hemorrhage from a ruptured cyst or a displaced kidney stone.
Genetic studies can confirm the diagnosis by identifying mutations in the relevant chromosomes associated with PKD.
Currently, there is no cure for polycystic kidney disease (PKD), so treatment focuses on relieving symptoms and preventing complications.
If PKD progresses to kidney failure (end-stage renal disease), dialysis or a kidney transplant becomes necessary.
Note: Using a twin or related donor may not be feasible due to the genetic nature of the disease.
Show more